本帖最后由 荷花池荒岛 于 2014-8-11 11:35 编辑
The Landscape of EGFR Pathways and Personalized Management of Non-small-cell Lung Cancer
http://www.medscape.com/viewarticle/740715_1
Abstract and Introduction
Abstract
Two classes of anti-EGF receptor (EGFR) agents, monoclonal anti-EGFR antibodies and small-molecule EGFR tyrosine kinase inhibitors, have been used for the treatment of non-small-cell lung cancer (NSCLC). However, only a subset of patients will benefit from EGFR-targeted therapy. The discovery of biomarkers that select the appropriate patients for the therapy and predict the responses to the therapy is urgently needed. Molecular genetic analyses provide new insights into EGFR pathway alterations and demonstrate promise for predicting the clinical outcome of patients with NSCLC. In this article, we summarize the latest available knowledge on the clinical impact of EGFR mutations, gene copy number, EGFR overexpression, phosphorylation expression and the alteration of the EGFR pathway downstream factors in predicting the response to EGFR-targeted therapy in NSCLC patients. The role of KRAS and BRAF mutations and ALK rearrangement in lung cancer-targeted therapy, are also reviewed.
Introduction
Non-small-cell lung cancer (NSCLC) accounts for approximately 80% of lung cancers and is one of the leading causes of cancer death in North America. It is often diagnosed at an advanced stage, with only 30–40% of metastatic NSCLC patients surviving for 12 months.[1–3]
Surgery is the most effective treatment for NSCLC; however, it is usually reserved for patients whose tumors are confined to the primary site and who have no or minimal lymph node involvement – a small portion of NSCLC cases. In addition, many patients who undergo curative surgery will later develop recurrence. Platinum-based chemotherapy is the mainstay of treatment in advanced or recurrent NSCLC, but the results of the treatment are far from encouraging.[1]
Approximately 85% of all primary lung cancers are NSCLCs, which are classified into three major histologic types: adenocarcinoma, squamous cell carcinoma and large-cell carcinoma.[4] Adenocarcinoma is the most frequent histologic type of NSCLC in both genders in many parts of the world (Figure 1). It accounts for approximately 40% of lung cancers and is usually found in the periphery of the lungs.[1] The most common histologic subtypes of adenocarcinoma are papillary (37%), acinar (30%), solid (25%) and bronchioloalveolar (7%).[5] Squamous cell carcinoma accounts for approximately 25–30% of all lung cancers and is often linked to a history of smoking. This cancer tends to occur in the hilar region of the lungs near the bronchus. Large-cell carcinoma accounts for approximately 10–15% of lung cancers and may appear in any part of the lung. It tends to grow and spread quickly and carries a poor prognosis.
Cigarette smoking remains the principal cause of lung cancer. It is estimated that 85–90% of all lung cancer patients have smoked cigarettes at some time.[6] As such, 87% of lung cancer deaths are thought to result from smoking. The US Environmental Protection Agency reports that radon is the second leading cause of lung cancer after cigarette smoking and is the leading environmental cause for nonsmokers. The risk of developing NSCLC from radon is much higher in people who smoke than in those who do not. Workplace exposure to asbestos fibers is another important, but less common, risk factor for lung cancer.[7–9]
In recent years, attention has turned to the role that the EGF receptor gene (EGFR) plays in tumorigenesis and its utility as a target for therapy. Biomarkers that can reliably predict which patients may benefit from anti-EGFR therapy are urgently needed. Pathologists will play a central role in the process to determine suitable testing and interpretation of the test results. In this article, we summarize the impact of EGFR alterations in predicting response to anti-EGFR therapies and discuss currently proposed technologies and their potential clinical implications.
Overview of EGFR & NSCLC
The EGFR and members of its family play an important role in carcinogenesis through their involvement in the modulation of cell proliferation, apoptosis, cell motility and neovascularization.[10] EGFR alterations have been implicated in the pathogenesis and progression of many malignancies.[11–14] Although the exact molecular pathways by which the mutant receptors lead to carcinogenesis are not completely understood, it is clear that mutant variants of EGFR have enhanced tyrosine kinase (TK) activity.
The presence of activating mutations in EGFR was initially reported in 2004.[12,15,16] Various groups also found amplification and overexpression of EGFR.[17–21] Clinical and pathological factors such as female gender, never having smoked, East Asian ethnicity and adenocarcinoma or bronchioloalveolar histology, correlated with objective responses to single-agent TK inhibitor (TKI) therapy in NSCLC and also with the presence of somatic EGFR mutations.[10,11,15,22,23] EGFR mutations rarely occurred in squamous cell carcinoma, large-cell carcinomas or adenocarcinomas with KRAS mutations.[5,12,15,20,22–31] Of great benefit to researchers in evaluating possible contributing mutations in NSCLC and acquired resistance to targeted therapies is the Catalogue of Somatic Mutations in Cancer (COSMIC[201]). COSMIC is designed to store and display somatic mutation information and related details relating to human cancers; it is a very useful tool for both researchers and clinicians. As more advanced molecular techniques reveal further molecular mutations, centralized databases such as COSMIC will allow clinicians and researchers to stay abreast of currently available information.
Lung adenocarcinomas frequently possess EGFR mutations and frequently exhibit increased EGFR copy number.[32] A study of 334 cases of lung adenocarcinomas using PCR-based assays to detect deletions within exon 19 and the L858R mutation in exon 21 of the EGFR gene found that 23% of these tumors contained a mutation. Of those, 71% were exon 19 deletions and 29% comprised the L858R mutation in exon 21.[32] In addition, EGFR amplification, defined as greater than five EGFR signals per nucleus by FISH, was detected in 52% of EGFR-mutated tumors, but in only 6% of those lacking the mutations. EGFR mutations were present in 26% of 86 bronchioloalveolar carcinomas.[24] It appears that EGFR mutations occur much less frequently in squamous cell carcinoma than in adenocarcinoma, with a reported incidence of 0–14%.[24,33] The third type of NSCLC, large-cell carcinoma, harbors EGFR mutations very rarely, if ever.[23,34] Marchetti et al. investigated a series of 31 large-cell carcinomas and found no EGFR mutations in any case.[24]
Motoi et al. reported that EGFR mutations are particularly frequent in adenocarcinoma of the papillary subtype.[5] In a group of NSCLC patients, 13 out of 36 (35%) papillary cancers harbored EGFR mutations, in contrast to three out of 63 nonpapillary cancers (5%). Kim et al. found that papillary subtype is a significant predictor of response to gefitinib in lung adenocarcinoma, although they did not link the incidence of response to the EGFR mutation status.[35]
The clinical implications of EGFR overexpression have been studied extensively, but the results are inconclusive thus far. Recent use of phosphor-specific antibody has facilitated analysis of the correlation between phosphorylation and EGFR mutation status. In a study of 218 cases of NSCLC, McMillen et al. correlated EGFR expression status with mutation status.[36] Phosphorylation at Y1045 was noted in 52% of cases, 71% of which exhibited the presence of an EGFR mutation. Phosphorylation of Y1068 was seen in 55% of cases, but it was present in 73% of those cases with an EGFR mutation. The data demonstrate that among Chinese patients, immunohistochemical detection of p-1045 and p-1068 expression predicts EGFR mutations.
Activation mutations identified within the kinase domain of the EGFR gene led researchers to propose and develop therapeutic strategies targeting EGFR TK. Therapeutic strategies targeting the EGFR pathway offered exciting new options for the treatment of NSCLC. EGFR alterations have prompted the development of two classes of anti-EGFR agents: monoclonal anti-EGFR antibodies (e.g., cetuximab and panitumumab) and small-molecule TKIs of EGFR (e.g., gefitinib and erlotinib, among others). According to large-cohort Phase III clinical trials,[37–40] the response rates range from 15 to 37.5%. Clinical trials were initiated that employed novel agents targeting the EGFR TK. The results of these clinical trials indicated that many of the tumors harboring mutant EGFR are highly sensitive to EGFR TKIs, with 10–30% demonstrating a significant clinical response.[15,41]
Summary
Constitutive activation of EGFR TK in NSCLC is associated with carcinogenesis of NSCLC.
The incidence of EGFR mutations in NSCLC is dependent upon gender, smoking history, tumor type and ethnic background.
Female gender, zero or very light smoking history, East Asian ethnicity, adenocarcinoma or bronchioloalveolar histology correlates with objective responses to single-agent TKI therapy in NSCLC.
EGFR Pathway Alteration & Implications
EGFR, located at chromosome 7p12, spans approximately 200 kb and contains 28 exons. It is a member of the ErbB family of four closely related TK receptors: EGFR (ErbB1), HER2/c-neu (ErbB2), HER3 (ErbB3) and HER4 (ErbB4).[42,43] EGFR is activated by binding of its specific ligands. Structurally, EGFR is composed of an N-terminal extracellular ligand-binding domain, a transmembrane lipophilic segment, and a C-terminal intracellular region containing a TK domain. Multiple ligands that bind and activate EGFR have been described, including EGF and TGF-α. Upon ligand binding to EGFR, the receptors form homo- or hetero-dimers, which activate their intrinsic intracellular protein TK. The ligand binding-induced dimerization results in cross-autophosphorylation of key tyrosine residues in the cytoplasmic domain, which function as docking sites for downstream signal transducers.[44] This activation of EGFR results in initiation of signaling cascades involving several downstream pathways.[4,26,45–50] Through its influence on these pathways, EGFR induces a number of crucial cellular responses, such as proliferation, differentiation, motility and enhanced cell survival (Figure 2).[50–53]

Figure 2.
EGFR and its signaling pathway. Structurally, an EGF receptor (EGFR) monomer is composed of an extracellular domain consisting of two ligand-binding subdomains: a transmembrane lipophilic segment and an intracellular region containing tyrosine kinase domains that occupy exons 18–24. The binding of ligands to EGFR results in autophosphorylation of key tyrosine residues in the cytoplasmic domain and activation of its intrinsic, intracellular protein tyrosine kinase activity. EGFR activation results in initiation of signaling cascades. These function to further modulate cell proliferation and survival through two downstream intermediate pathways: the PI3K–AKT–mTOR pathway and the RAS–RAF–MEK–MAPK pathway. These two intermediate pathways influence several key aspects of the cell cycle that include cell proliferation, apoptosis, migration and survival, and more complex processes such as angiogenesis.
P: Phosphorylation.
One of the major molecular alterations in the carcinogenesis of NSCLC is the activation mutation of EGFR. The mechanisms that regulate EGFR expression, such as epigenetic alteration and aberrant transcription factors, have been studied, but with inconclusive results. The significance of miRNA-128b was reported by Weiss et al., who found loss of miRNA-128b in two out of three NSCLC cell lines and in tumors from 55% of NSCLC patients.[54] miRNA-128b directly regulated EGFR expression, and loss of miRNA-128b correlated with better tumor responsiveness to gefitinib treatment and improved survival (23.4 vs 10.5 months).[54]
A strong genetic association with particular germline mutations has been shown to influence the susceptibility to EGFR TKIs (i.e., those that confer mutations in EGFR signaling). Liu et al. found that the frequencies of the -216T and CA-19 alleles are significantly higher in patients with any mutation, in particular in those with exon 19 microdeletions.[55] The -216T allele is preferentially amplified in human lung cancer specimens and cancer cell lines. These results suggest that the local haplotype structures across the EGFR gene may favor the development of carcinogenesis and thus significantly confer risk to the occurrence of EGFR mutations in NSCLC, particularly the exon 19-microdeleted cases.[55] A novel germline transmission of the EGFR mutation V843I in a family with multiple members with lung cancer has been reported.[56] The proband was a 70-year-old woman who had multiple adenocarcinomas with EGFR mutations. These observations suggest that germline EGFR V843I mutation may result in altered EGFR signaling in cases of multicentric adenocarcinoma, bronchioloalveolar carcinoma and atypical adenomatous hyperplasia, and may also play a role in the development of lung cancer in multiple family members.[56]
EGFR plays a key role in the growth and survival of many solid tumor types.[21,57] Mutations affecting EGFR activity or expression can result in cancer.[58] The EGFR TK modulates cell proliferation and survival through two downstream intermediate pathways: the PI3K–AKT–mTOR pathway and the RAS–RAF–MEK–MAPK pathway.[59] These downstream cell signaling pathways influence several critical cellular processes, including cell proliferation, apoptosis, migration and survival. They are also involved in more complex processes such as angiogenesis and tumorigenesis. Studies on EGFR oncogene activation have been focused on gene mutations, DNA copy number alterations, protein expression alterations and genetic alterations of downstream signaling molecules.
Results from a Phase III trial evaluating the EGFR TKI, gefitinib, indicated that approximately 10% of patients responded to the therapy, and no survival benefit was observed.[30] Follow-up analysis identified mutations in the TK domain of EGFR in eight out of nine responders, whereas no mutations were detectable in seven patients who did not respond to gefitinib therapy.[12]
The most widely used EGFR TKIs are gefitinib and erlotinib. These agents are reversible inhibitors that compete with ATP at the active site of the TK receptor domain.[6] They are primarily used in patients who have failed platinum-based chemotherapy.[60] In a randomized Phase III study, erlotinib significantly improved median survival from 4.7 (placebo) to 6.7 months in patients with NSCLC who had previously failed one or two chemotherapy regimens.[61] Gefitinib improved disease-related symptoms in heavily pretreated symptomatic patients with NSCLC.[30] However, in the Phase III Iressa Survival Evaluation in Advanced Lung Cancer (ISEL) trial, which included pretreated patients with recurrent disease, gefitinib failed to demonstrate a survival benefit in the overall unselected patient population compared with placebo-treated controls.[62] Recently, however, results from the randomized Phase III IRESSA NSCLC Trial Evaluating Response and Survival against Taxotere (INTEREST) indicate that gefitinib is not inferior to docetaxel in terms of overall survival, suggesting that this TKI may also be a viable option for previously treated patients with advanced NSCLC.[63]
The data from 222 publications indicate that EGFR mutations are predictive of patient response to single-agent EGFR TKI treatment in advanced NSCLC.[64]
EGFR Mutations
The EGFR mutations responsible for the constitutive activation of receptor TK are also most frequently associated with sensitivity to EGFR TKIs.[65] These mutations are associated with response rates of >70% in patients treated with either erlotinib or gefitinib.[28,66]
Receptors containing different mutations appear to have different signaling properties, but most mutations seem to affect the ATP-binding cleft, which is also where targeting TKIs bind (Figure 3).[49]

Figure 3.
Mechanism of EGFR-activating mutations and EGFR-targeted therapy. (A) EGFR mutations render EGFR tyrosine kinase constitutively activated. Activated EGFR phosphorylates key tyrosine residues (P) in the tyrosine domain, which initiates downstream effectors. (B) At present, two categories of agents are used for inhibiting EGFR signaling: humanized antibodies and small-molecule TKIs. Antibodies inhibit ligand-dependent activation of EGFR by blocking the ligand-binding site and preventing activation. TKIs block the magnesium–ATP-binding pocket of the intracellular tyrosine kinase domain, further inhibiting autophosphorylation. This inhibition disrupts tyrosine kinase activity and abrogates intracellular downstream signaling.
TKI: Tyrosine kinase inhibitor.
In vitro studies have demonstrated that mutant EGFR has enhanced TK activity, leading to a greater sensitivity to anti-EGFR inhibition. As mentioned previously, the four most common mutations seem to be those most closely associated with TKI sensitivity. The discovery that many objective responders to TKIs harbored EGFR mutations in exons 19 and 21 was a major breakthrough in patient selection for EGFR targeting therapy.[67,68] The most frequent mutation, located in exon 19, eliminates four amino acids – leucine, arginine, glutamic acid and alanine – downstream from the lysine residue at position 745.[67,69–72] Patients with an EGFR mutation who were treated with TKI had much higher response rates and longer progression-free survival than those without a mutation (Table 1).[69]
Both retrospective and prospective studies have demonstrated that NSCLC patients carrying the described EGFR gene mutations have a significantly higher response rate to gefitinib and/or erlotinib compared with patients with wild-type EGFR.[12,15,16,28,67,68,73–76] Some patients experienced rapid complete or partial responses that were durable.[26] The discovery of somatic mutations in EGFR that correlated with sensitivity to TKIs identified a plausible and reproducible explanation for these observations.
The most commonly used methods to detect mutations are direct sequencing and real-time PCR.[73,77] Other methods include single-strand conformational polymorphism analysis[78,79] and high-resolution melting amplicon analysis.[17,80] Scorpion ARMS® (QIAGEN, Germany), a multitargeted real-time PCR detection kit, allows the detection of the most prevalent somatic mutations in the EGFR that are common in human cancers. The high sensitivity and specificity of the kit permits the detection of mutations against a background of wild-type genomic DNA. The kit uses DxS Scorpions® (QIAGEN) technology to detect exon 19 deletions and mutations in exons 19–21 (T790M, L858R, L861Q, G719X and S768I) and any one of three insertions into exon 19 (2307_2308ins9, 2319_2320insCAC and 2310_2311insGGT). Relative to the direct sequencing method, the other two techniques allow for the rapid detection of EGFR mutations with high sensitivity and specificity. However, confirmation of mutations via direct sequencing is necessary.[24,80,81] Standardization is essential for the clinical application of EGFR mutation tests. However, at present, there is no official guideline for these EGFR mutation tests. The number of mutation sites that are needed in the testing protocol still remains to be established. Large-scale clinical trials are also needed.
Jackman et al. studied 223 chemotherapy-naive patients with advanced NSCLC.[28] Sensitizing EGFR mutations were associated with a 67% response rate, with a time to progression (TTP) of 11.8 months and overall survival of 23.9 months. Exon 19 deletions were associated with a longer median TTP and overall survival compared with L858R (exon 21) mutations. Wild-type EGFR was associated with poor outcomes (response rate: 3%; TTP: 3.2 months), irrespective of KRAS status. EGFR genotype was more effective than clinical characteristics at selecting appropriate patients for consideration of first-line therapy with an EGFR TKI.
Studies indicate that more than 75% of patients responsive to TKI therapy have activating mutations in EGFR.[13,77] However, some rare types of EGFR mutations can confer resistance to EGFR-targeted therapies after treatment with TKIs when combined with the common activating mutations.[82–85] Clinically, patients with EGFR exon 20 mutations do not respond to gefitinib.[72] Moreover, the appearance of a secondary mutation in exon 20 (T790M) accounts for approximately 50% of acquired drug resistance.[77,86] Screening for the emergence of such mutations in circulating tumor cells from the blood of patients during the course of treatment may allow earlier identification of acquired resistance.[83,87]
Results of some preclinical studies suggest that the clinical benefit observed with EGFR TKIs is not restricted to those patients harboring EGFR mutations. This may be due to molecular factors outside of gene mutations. EGFR amplification and receptor/ligand overexpression, both of which allow for a 'gain of function' to occur, are implicated in creating a scenario of EGFR dependence that causes the sensitivity to single-agent EGFR inhibitors.[44,88] However, the data from the IRESSA Pan-Asia Study (IPASS) clearly demonstrate that patients with increased EGFR copy numbers and no EGFR mutations do not benefit from EGFR TKIs.[89]
EGFR Copy Number Alterations
EGFR is frequently over-represented or amplified in NSCLC, which is commonly associated with EGFR overexpression.[90] Increased EGFR copy numbers may result from gene amplification or polysomy of chromosome 7. The incidence of EGFR amplification ranges from 12 to 59%, depending on the patients selected and the technology used.[64,88,91,92] Some, but not all, studies have revealed that positive EGFR amplification is associated with significantly better survival after treatment with a TKI.[88,93] Gain of EGFR copy number has been consistently associated with a favorable outcome after EGFR TKI therapy; it has also been proposed to be a potential biomarker of TKI responsiveness.[91,94]
Somatic EGFR mutations consistently correlate with improved response rates; by contrast, the results of studies investigating EGFR copy number as a predictor of response to TKIs have been inconsistent.[25,95] Overall, EGFR mutations seem to have higher sensitivity and specificity for predicting response to TKIs than EGFR copy number gain status.[64] High copy numbers of EGFR have been detected in approximately 30% of NSCLC patients using FISH, and are reportedly associated with better responses to TKI therapy,[73,96] although the EGFR mutation status of those cases was unclear. Approximately 70% of patients with EGFR copy number gain also had EGFR somatic mutations, a fact that clouds the true significance of EGFR copy number gain. IPASS demonstrated that EGFR mutation was the strongest predictor of improved progression-free survival. There was a high degree of overlap between EGFR mutation positivity and high EGFR gene copy numbers: of 245 patients with high EGFR copy numbers whose EGFR mutation status was also known, 190 (78%) were also EGFR mutation-positive. This suggests that the improved outcome in high EGFR copy number patients is being driven by the EGFR mutation-positive overlap.[89] Some researchers have suggested that high EGFR copy numbers can be used as a predictive biomarker for response and survival benefit in patients with NSCLC who receive EGFR TKI therapy.[88] However, the data from the IPASS trial clearly demonstrate that patients with increased EGFR copy numbers and no EGFR mutations do not benefit from EGFR TKIs.[89] Hirsch et al. suggested that although EGFR mutations and high copy numbers are both predictive of response to erlotinib in NSCLC, EGFR copy number was a more powerful predictor of differential survival benefit from erlotinib.[88]
There are several methods for detecting and determining EGFR copy number and dosage, including FISH,[73,88,97] chromogenic in situ hybridization[92,98] and real-time quantitative PCR.[20,99,100] When EGFR copy number was measured by PCR, it was found that increased EGFR copy number was significantly associated with prolonged survival, indicating a potential prognostic value of EGFR copy number.[64,101] The patients with EGFR gain demonstrated a higher disease control rate (67 vs 26%), longer TTP (9.0 vs 2.5 months) and prolonged survival time (18.7 vs 7.0 months).[64] It is noteworthy that EGFR copy number is used as a predictor for response to TKI therapy largely because it is correlated with EGFR mutations – EGFR mutations are the best predictors. Hirsch et al. investigated 229 chemotherapy-naive patients with advanced-stage NSCLC in a Phase II clinical trial.[88] Among the 76 patients analyzed by FISH, 59.2% had increased EGFR copy numbers, as indicated by four or more gene copies per cell in >40% of the cells. Response was higher in EGFR-amplified patients (45%) versus the EGFR-unamplified patients (26%). Those patients with EGFR amplification had a median progression-free survival time of 6 months compared with 3 months for patients without amplification. Median overall survival was 15 months for the EGFR-amplified group, while it was only 7 months for patients without amplification.[102]
Despite a majority of studies demonstrating that high EGFR copy number correlates with better response and increased survival in NSCLC patients treated with EGFR TKIs, debate remains about its true predictive value. Some studies suggest that when compared with EGFR mutations, EGFR gene copy number is a less sensitive and less specific marker and may not be considered clinically suitable for patient selection.[64] Douillard et al. also reported that EGFR mutation demonstrates greater predictive power than EGFR copy number in therapy response and progression-free survival.[103] Further studies are necessary to resolve these discrepant findings.
EGFR Overexpression
The clinical implications of EGFR overexpression have been studied extensively. However, the results have been inconclusive thus far. Immunohistochemistry-based assays measuring EGFR expression could not reliably predict the response to EGFR TKI therapy. Overexpression of EGFR has been demonstrated in 40–80% of cases of NSCLC and has been associated with a poor prognosis.[104–106] The initial assumption was that EGFR antibodies would be more effective in tumors with robust overexpression of EGFR. However, early clinical studies were unable to demonstrate a distinct correlation between EGFR expression and the likelihood of response to EGFR inhibition with targeted antibodies.[107] In addition, studies suggest that immunohistochemistry-based assays measuring EGFR expression do not serve as reliable predictors of response to cetuximab therapy.[108]
Increased response rates after treatment with a TKI have been demonstrated in patients with positive EGFR immunostaining in some studies, but not in others.[104,105,109,110] In multivariate analyses, EGFR expression level was associated with an objective response or adverse prognosis in NSCLC.[93,110] Several investigations into the prognostic significance of EGFR expression revealed no association with survival benefit.[102,105,111] Therefore, EGFR overexpression by itself is not prognostic of survival in NSCLC. It has been suggested that the nonoptimized cut-off value for EGFR-positive immunostaining and/or lack of standardization in staining procedures and guidelines may explain the discordance among these studies.[111]
EGFR Mutation-specific Antibodies
Since the use of EGFR overexpression as a prognostic marker has largely been unproductive, considerable efforts have been made to develop antibodies that react specifically with the mutant form of EGFR. Cell Signaling Technology, Inc. (MA, USA) has developed two mutant-specific antibodies against the most common mutant forms of EGFR: the 15-base pair/5-amino acid deletion (E746-A750del) in exon 19 and the L858R point mutation in exon 21.[112] Yu et al. investigated EGFR genotypes of 40 NSCLC tumor samples by immunohistochemistry with these antibodies and confirmed the immunohistochemistry results by DNA sequencing.[112] Detection of mutant EGFR by these two antibodies was performed by western blotting, immunofluorescence and immunohistochemistry. The sensitivity and specificity of these antibodies in a 340-sample panel of paraffin-embedded NSCLC tumors was 92 and 99%, respectively, compared with direct sequencing and mass spectrometry-based DNA sequencing. These results demonstrate that mutation-specific antibodies provide a rapid, sensitive, specific and cost–effective method to identify lung cancer patients who may respond to EGFR-targeted therapies. Brevet et al. evaluated the two mutation-specific monoclonal antibodies for the detection of EGFR mutations by immunohistochemistry on 218 paraffin-embedded lung adenocarcinomas.[112] The EFGR L858R mutant antibody showed a sensitivity of 95%, a positive predictive value of 99% and a specificity of 76%, with a positive cut-off of (2+).[113] A positive threshold of (2+) will effectively reduce the false-positive rate and enhance the predictive power of immunohistochemistry assays to 100%, with a minimal reduction in sensitivity. The immunostaining scoring was based on cytoplasmic and/or membrane staining intensity as follows: (0+) = no staining or faint staining intensity in <10% of tumor cells; (1+) = faint staining in >10% of tumor cells; (2+) = moderate staining; and (3+) = strong staining. Therefore, immunohistochemistry using mutation-specific antibodies can be used to screen for patients who may be candidates for EGFR inhibitors.[113]
Phosphorylated Form of EGFR
Aberrant activation of EGFR is a recognized component of cancer development and progression.[59] In addition, recent data indicate that both EGFR mutations and the activation status of EGFR, defined by phosphorylation, might have a strong impact on the clinical course of NSCLC.[114] The two major EGFR signaling pathways, PI3K–AKT–mTOR and RAS–RAF–MAPK, mediate EGFR effects on cell proliferation and survival. The activation of these pathways is dependent on the phosphorylation status of the components. Investigations to date indicate that the major molecular alteration involved in the carcinogenesis of NSCLC is an activation mutation. The mechanisms that regulate EGFR expression, such as epigenetic alteration and aberrant transcription factors have been studied but are not yet conclusive.
Phosphorylation at tyrosine 845 in the kinase domain of EGFR may stabilize the activation loop, which maintains the receptor in an active state and provides a binding surface for substrate proteins.[115] Phosphorylation of two additional tyrosines, 1068 and 1173, mediates the direct binding of growth factor receptor-bound protein 2. Furthermore, tyrosine 1068 is involved in the activation of the MAPK signaling pathway.[116]
Detection of activated EGFR is conducted by using anti-phospho-EGFR antibodies directed at EGFR in its phosphorylated state. Phosphorylations in the carboxyl-terminus of EGFR play a key role in the recruitment of signaling molecules and activation of downstream signaling pathways.[115,117] In a study by Endoh et al., involving 97 NSCLC cases, patients with phospho-EGFR-positive tumors demonstrated a prolonged survival, although the follow-up period was relatively short.[117] Hijiya et al. investigated 21 cases of NSCLC for correlations between the presence of EGFR mutations and the EGFR phosphorylation status by immunohistochemistry with antibodies recognizing EGFR that was phosphorylated at tyrosine 992 and tyrosine 1173, respectively.[118] The mutation status of EGFR was strongly correlated with immunoreactivity for phosphorylated tyrosine 992, indicating a clear potential for using anti-phospho-EGFR antibodies as a surrogate marker of EGFR mutations and thus predicting the clinical response to tyrosine antagonist therapy.
The immunohistochemical evaluation of NSCLC with anti-phospho-EGFR antibodies is potentially useful in the clinical prediction of responsiveness to EGFR-targeted therapy. However, further testing and evaluation are needed to determine its true clinical implication.
EGFRvIII
The newly characterized EGFR mutant, EGFRvIII, results from an in-frame deletion of exons 2–7 of the coding sequence, which has been found to be generated by gene rearrangement or aberrant mRNA splicing.[119,120] The variant form has a deletion of 267 amino acids in the extracellular domain of normal EGFR, creating a unique epitope at the fusion junction. A number of functional differences between EGFRvIII and EGFR have been characterized.[120,121] Although EGFRvIII fails to bind EGF, its intracellular TK is constitutively activated, allowing the receptor to undergo tyrosine autophosphorylation.[122,123] These studies provide further evidence that EGFRvIII expressed in NSCLC is phosphorylated and, hence, activated. The data suggest that the sustained activation of EGFRvIII may play a role in the pathogenesis of NSCLC and, therefore, EGFRvIII is a potential therapeutic target for NSCLC.[114] Antibodies directed to this tumor-specific variant of EGFR provide an alternative targeting strategy. It has been demonstrated that systemic treatment of mice bearing tumors expressing EGFRvIII with monoclonal antibodies specific for EGFRvIII inhibited tumor growth and extended animal survival.[124,125] Antibodies that have an affinity for EGFRvIII, but without an affinity to wild-type EGFR, provide an alternative tool for detecting this mutation variant.[126]
The role of EGFRvIII mutations in the pathogenesis of NSCLC is unclear. Reported incidences of EGFRvIII mutation in NSCLC vary from 0 to 42%.[127,128] These differences may be due to differences in the tumor composition (histological type) or to technical considerations, such as the threshold for the result interpretations. Studies using immunohistochemical assays with EGFRvIII mutant-specific antibodies suggest that this mutation is present in multiple other tumor types and is not exclusive to NSCLC.[114,129] However, owing to the large size and complex genomic structure (28 exons spanning ~190 kb) of EGFR and its large intron 1 (123 kb), where genomic deletions frequently occur, it has been difficult to assess and verify the existence of the EGFRvIII mutations at the genomic level.[127] To evaluate the clinical impact of EGFRvIII in NSCLC, Okamoto et al. investigated EGFRvIII expression in 76 cases of NSCLC by immunohistochemistry, using a monoclonal antibody specific for this EGFR variant. EGFRvIII expression was found in 39% (30/76) of NSCLC; however, genetic analysis of EGFRvIII mutations only generated a 3% positive rate compared with the 39% immunopositivity rate.[114] Okamoto et al. found that EGFRvIII was also observed in several normal tissue components of the lung, which raised the question of the clinical implications for this detection methodology.[114]
Studies of small-molecule TKIs have demonstrated clinical responses in NSCLC patients whose tumors bear EGFR kinase domain mutations. However, the efficacy of these inhibitors against tumors with the EGFRvIII mutation remains unclear. Ji et al. determined that EGFRvIII mutations were present in 5% (3/56) of human squamous cell lung carcinomas, but found no EGFRvIII mutations in a large cohort of human lung adenocarcinomas (0/123).[127] In their study, EGFRvIII-bearing tumors seemed relatively resistant to some TKIs, but responsive to others.[130] In an in vivo system, treatment with an irreversible EGFR inhibitor, HKI-272, dramatically reduced the size of EGFRvIII-driven murine tumors within 1 week.[126] A total of 7 days of erlotinib treatment led to an average reduction of 45% in tumor volume in the three treated mice. By contrast, those treated with HKI-272 demonstrated a reduction of 88%. The Ba/F3 cells, transformed with the EGFRvIII mutant, were relatively resistant to gefitinib and erlotinib in vitro, but sensitive to HKI-272, suggesting that TKI treatment is potentially efficacious for cancers harboring the EGFRvIII mutation.
Summary
Current studies of alterations of the EGFR pathway have been focused on gene mutations, gene copy-number alterations, protein expression alterations and downstream genetic alterations.
Four activating mutations – exon 18 (G719A/C), exon 21 (L858R and L861Q), and in-frame deletions in exon 19 – are the dominant mutations present in NSCLCs.
Patients with an EGFR mutation, who were treated with TKIs, had much higher response rates and longer progression-free survival than patients without EGFR mutations who had the same treatment.
Acquisition of a new mutation in exon 20 can confer resistance to TKI treatment.
Overexpression of EGFR has been found in 40–80% of cases, but its usefulness as a predictive marker remains controversial.
Sustained activation of EGFRvIII is implicated in the pathogenesis of squamous cell carcinoma and, thus, EGFRvIII is a potential therapeutic target in this subset of NSCLCs.
EGFR-targeted Therapy & Mechanisms of Resistance
There are two major approaches for inhibiting EGFR signaling: to prevent ligand binding to the extracellular domain with a monoclonal antibody and to inhibit the intracellular TK activity with a small molecule. Use of the latter approach was the first method to be attempted clinically.[131] The EGFR TKIs are reversible competitive inhibitors of the TK domain of EGFR that bind to its ATP-binding site. Somatic activating mutations of the EGFR gene, increased gene copy number and certain clinical and pathological features have been associated with dramatic tumor responses and favorable clinical outcomes with these agents in patients with NSCLC. The majority of these patients inevitably acquire resistance to EGFR TKIs. Recent data indicate that a secondary mutation, such as T790M, expression of HGF, PTEN and/or early growth response-1 and changes in the epithelial-to-mesenchymal transition, were associated with EGFR TKI resistance. Uramoto et al. found that strong expression of HGF was detected in six out of eight specimens with the T790M mutation.[132] Three out of eight cases (38%) demonstrated a loss of PTEN in samples with the T790M mutation. A loss of early growth response-1 was detected in two out of seven cases (29%), including one tumor without PTEN. Four out of seven cases (57%) demonstrated positive expression of phosphorylated Akt. A change in the epithelial-to-mesenchymal transition status between pre- and post-treatment was observed in four out of nine cases (44%). These results suggest that alterations in gene or protein expression can account for all mechanisms by which tumors acquire resistance to EGFR TKIs.[132] This phenomenon suggests the existence of complicated relationships between acquired resistance-related genes.
Somatic activating mutations in EGFR are identified in a subset of NSCLC that responds to TKIs. As noted previously, acquisition of drug resistance has been linked to a specific secondary somatic mutation, EGFR T790M. Bell et al. described a family in which multiple members developed NSCLC associated with germline EGFR mutations of T790M.[25] These observations implicate altered EGFR signaling as a culprit in the genetic susceptibility to lung cancer in families with an increased incidence of NSCLC.
We propose an algorithm for molecular testing for patients with NSCLC (Figure 4). A stepwise approach, based on the frequency of specific mutations, is used to assess lung cancer patients for specific findings, which will allow proper therapeutic stratification for targeted therapy.
Figure 4.
Suggested algorithm for molecular testing for patients with non-small-cell lung cancer. A stepwise approach is used to test lung cancer patients according to the known frequencies of various mutations. Small-cell lung cancers are excluded from testing. NSCLC, which accounts for approximately 85% of all lung cancers, is tested for the presence of EGFR mutations. A positive test, found in approximately 20% of Caucasians and 40% of East Asians, predicts an 80% probability of response to EGFR TKI therapy. Nonmutated EGFR is found in approximately 80% of Caucasians and 60% of East Asians. These patients are further tested for EML4–ALK mutations. EML4–ALK mutations are found in only 3% of patients with NSCLC, but the mutation predicts a 53% probability of response to targeted therapy. Cases lacking EML4–ALK mutations may undergo additional testing.
EGFR: EGF receptor; Mu: Mutation; NSCLC: Non-small-cell lung cancer; SCC: Small-cell carcinoma; TKI: Tyrosine kinase inhibitor.
EGFR-targeted Therapy Approaches
Monoclonal Antibodies Monoclonal antibodies, such as cetuximab and panitumumab, are either chimeric mouse–human or fully humanized antibodies targeting the extracellular domain of EGFR and thereby inhibiting the binding of activating ligands to the receptor. This class of treatment inhibits ligand-dependent activation of EGFR and inhibits the downstream pathways, which cause cell cycle progression, cell growth and angiogenesis (Figure 3A). In addition, binding of the antibody initiates EGFR internalization and degradation, which leads to signal termination.[108,133] Fully humanized antibodies such as panitumumab, have a high affinity for EGFR and a longer half-life.[134] Although EGFR is frequently expressed in patients with NSCLC, the clinical efficacy of treatment with anti-EGFR antibodies is limited to only a subset of patients.
Tyrosine kinase inhibitors Tyrosine kinase inhibitors are synthetic small molecules that block the magnesium–ATP-binding pocket of the intracellular TK domain.[108] Several TKIs, such as gefitinib and erlotinib, are specific for EGFR, whereas others inhibit other receptors in addition to EGFR, such as HER2 and VEGF receptor 2. TKIs block ligand-induced receptor autophosphorylation by binding to the TK domain and disrupting TK activity, thereby abrogating intracellular downstream signaling (Figure 3B & Table 1).
Mechanisms of Resistance to EGFR-targeted Therapy
Acquired Resistance Caused by a Secondary Mutation Although EGFR mutations are associated with enhanced sensitivity to gefitinib and erlotinib, not all tumors that have activating mutations are associated with an enhanced response. The efficacy of EGFR TKIs is limited owing to either primary or acquired resistance after therapy. Most patients who initially respond to gefitinib and erlotinib eventually become resistant and experience progressive disease.
It is known that four mutations result in TKI drug sensitivity: point mutations in exon 18 (G719A/C) and exon 21 (L858R and L861Q), as well as in-frame deletions in exon 19, which eliminate four amino acids – leucine, arginine, glutamic acid and alanine – downstream of the lysine residue at position 745.[67,69–72] However, insertion mutations of exon 20 at D770–N771 were associated with EGFR TKI resistance.[60,135] This observation was confirmed in an in vitro model in which insertion mutations in exon 20 rendered transformed cells less responsive to EGFR TKIs compared with the sensitizing mutations of exons 19 and 21.[135]
Two established mechanisms of acquired resistance consist of additional mutations in the EGFR gene acquired during the course of treatment that change the protein coding sequence and amplification of other oncogene signaling pathways.[85,136–138]
Kobayashi et al. reported a gefitinib-resistant advanced NSCLC patient who had a relapse after 2 years of complete remission due to treatment with gefitinib.[86] The DNA sequence of EGFR at relapse revealed the presence of a second point mutation, resulting in a T790M mutation of EGFR. Structural modeling and biochemical studies showed that this second mutation led to gefitinib resistance.[86] The same mutation was confirmed by Pao et al. through molecular analysis of EGFR in patients with acquired resistance to gefitinib or erlotinib.[139] The gefitinib-resistant cases contain the same secondary mutation (T790M) in the kinase domain.[22] Codon 790 of EGFR is considered to be the 'gatekeeper' residue, which is an important determinant of inhibitor specificity in the ATP-binding pocket of EGFR.[108] Substitution of this residue in EGFR with a bulky methionine may cause resistance by steric interference with the binding of TKIs, including gefitinib and erlotinib.[86,139,140] This mutation may confer a survival advantage to the tumor and is probably selected for while the patient is receiving anti-EGFR TKI treatment.[25,84] These findings have led to the development of irreversible EGFR TKIs in an effort to effectively target this mechanism of resistance.[140]
The role of oncogenic activation of EGFR downstream effectors, such as KRAS, BRAF, PIK3CA and PTEN, in response to therapy is discussed extensively in a series of studies.[47,53] The RAS–MAPK and PI3K–AKT pathways are major signaling networks linking EGFR activation to cell proliferation and survival.[141,142] Mutations in these downstream effectors of EGFR signaling could lead to resistance to EGFR inhibitors.[136–138] The discovery of molecular aberrations, such as MET kinase amplification or mutations of EML4–ALK fusion, which causes constitutive activation of RAS–RAF–MEK, has provided further insight and validation into factors limiting the therapeutic efficacy of EGFR inhibitors.[46,143,144]
KRAS Mutations KRAS plays a key role in the EGFR signaling network. The KRAS proto-oncogene encodes KRAS G-protein, which plays a critical role in the RAS–MAPK signaling pathway downstream of many growth factor receptors, including EGFR.
One of the most important discoveries for the clinical management of colorectal carcinoma has been the association of mutations in KRAS and the efficacy of monoclonal antibodies targeting EGFR, such as panitumumab and cetuximab. Some tumors harbor somatic mutations in exon 2 of KRAS that compromise the hydrolysis of RAS-bound GTP to GDP, resulting in constitutive activation of the RAS pathway.[145] In the presence of a KRAS mutation, EGFR pathway activation cannot be significantly inhibited by cetuximab or panitumumab, which acts upstream of the KRAS protein, diminishing the efficacy of the agents.
An activating mutation of KRAS is present in 15–30% of NSCLC cases[26,146,147] and accounts for approximately 35–45% of TKI-nonresponsive cases.[148] Approximately 30% of lung adenocarcinomas contain activating KRAS mutations, which are associated with resistance to EGFR TKIs.[22] It is noteworthy that the presence of a KRAS mutation is common in NSCLC, but the occurrences of KRAS and EGFR mutations seem to be mutually exclusive.[4,27,149–152] EGFR and KRAS mutations are rarely if ever detected in the same tumor, suggesting that they may perform functionally equivalent roles in lung tumorigenesis.[58,153] However, there is growing evidence that coexistence of EGFR and KRAS mutations is possible,[27,151,154] although the frequency is low. Due to the limited number of cases, it is difficult to obtain conclusive results; however, the available data suggests a negative association between EGFR/KRAS mutation and EGFR TKI responsiveness.[27,151,154]
It remains unclear whether assessment of KRAS mutation status will prove to be clinically useful with regard to anti-EGFR therapy.[50] Although an association between the presence of a KRAS mutation and lack of response to EGFR TKIs has been observed, it remains indeterminate whether this association is clinically relevant with respect to progression-free and overall survival. Investigations of KRAS mutation status as a negative predictor of outcome after anti-EGFR therapy have been undertaken, but small sample sizes due to low prevalence of KRAS mutations have limited the power of such studies. Some investigators have reported that KRAS mutation is a negative predictor of response to anti-EGFR monoclonal antibodies and also an important mechanism of resistance to TKIs in NSCLC.[26] Unlike colorectal cancer, KRAS mutations do not seem to identify patients who do not benefit from anti-EGFR monoclonal antibodies in NSCLC.
KRAS mutations are almost exclusively detected in codons 12 and 13 of exon 2, which may result in EGFR-independent intracellular signal transduction activation. In a study by Eberhard et al., EGFR exons 18–21 and KRAS exon 2 mutations were investigated via sequencing in tumors of 274 patients.[27] KRAS mutations were present in 21% of tumors, which were associated with significantly decreased TTP and survival in patients treated with erlotinib plus chemotherapy. Others have reported that KRAS mutation status did not impact EGFR TKI therapy.[28] In a study that included 223 chemotherapy-naive patients with advanced NSCLC treated with erlotinib or gefitinib monotherapy, EGFR mutations were associated with a 67% response rate. Wild-type EGFR was associated with poorer outcomes, regardless of KRAS mutation status.[28]
A study by Wang et al. utilizing PCR-restriction fragment length polymorphism analysis investigated the KRAS mutations in codons 12 and 13 in 273 NSCLC cases.[155] Of the 120 patients who received EGFR TKI therapy, only 5.3% (one out of 19) of the patients with a KRAS mutation demonstrated a response compared with a 29.7% response rate for patients lacking a KRAS mutation.[152] Furthermore, the median progression-free survival time of patients with a KRAS mutation was 2.5 months compared with 8.8 months for patients with wild-type KRAS.
A meta-analysis by Linardou et al. provided empirical evidence that somatic mutations of the KRAS oncogene are highly specific negative predictors of response to single-agent EGFR TKIs in advanced NSCLC.[148] Among 17 publications, 165 out of 1008 (16%) NSCLC patients presented with KRAS mutations. The presence of KRAS mutations was significantly associated with an absence of response to TKIs in these patients.
Having an intact KRAS is necessary, but not sufficient, to derive benefit from EGFR inhibition, and additional mechanisms of resistance to EGFR inhibitors exist.
KRAS testing scenarios in the management of NSCLC are summarized in Figure 5. The incidence of KRAS mutations in NSCLC is reportedly up to 20%.[156] In the subset of tumors with KRAS mutations, less than 3% also contain an EGFR mutation, and the remaining 97% of tumors with KRAS mutations have wild-type EGFR. Both KRAS/EGFR double mutations and wild-type EGFR are associated with nonresponsiveness to EGFR-targeted therapy.
Figure 5.
KRAS testing scenarios in the management of non-small-cell lung cancer. NSCLC accounts for approximately 85% of all lung cancers. The incidence of KRAS mutations in NSCLC is reportedly up to 20%. In the subset of patients with KRAS mutation, less than 3% of the tumors also contain an EGFR mutation; the remaining 97% have wild-type EGFR. Both KRAS/EGFR double mutations and wild-type EGFR are associated with nonresponsiveness to EGFR-targeted therapy. Of the 80% of NSCLCs that do not have KRAS mutations, approximately 20% harbor EGFR mutations, which are associated with an 80% likelihood of response to EGFR TKI therapy.
EGFR: EGF receptor; Mu: Mutation; NSCLC: Non-small-cell lung cancer; TKI: Tyrosine kinase inhibitor.
lung without KRAS and EGFR mutations.[160] Of these, ten adenocarcinomas with the BRAF-V600E mutation were identified. BRAF mutations were reported more frequently in micropapillary lung adenocarcinoma.[161] De Oliveira Duarte Achcar and colleagues analyzed the clinical and molecular profile of 15 primary micropapillary adenocarcinomas and found BRAF mutations in three cases (20%). The BRAF-V600E mutation-bearing tumors had a slight female predilection (6:4 female:male). The elderly patients were found to have a greater than expected incidence of intralobar satellite nodules and N2 node involvement.[161] The adenocarcinomas were largely of mixed type, with a high incidence of papillary (80%) and lepidic growth (50%). However, due to the relatively small sample size, it is yet to be determined whether BRAF mutant tumors represent a distinct subset of lung adenocarcinoma.[162]
Mutations in BRAF have been shown to impair responsiveness to panitumumab or cetuximab in patients with colorectal carcinomas. This initial retrospective work was performed on a cohort of 132 patients.[163] The results showed that none of the patients who experienced a response displayed BRAF mutations, whereas 11 of 79 nonresponders (14.0%) carried a BRAF-V600E allele.
Nevertheless, BRAF mutations are rarely detected in NSCLC when compared with KRAS mutations.[162,164,165] A total of 80% of the reported mutations are located within the kinase domain of BRAF.[159] Brose et al. identified activating BRAF mutations in five out of 292 cases (1.7%) of NSCLC. Among these mutations, three were found in exon 11 and two in exon 15.[164] It has been proposed that mutations in BRAF, a downstream signaling molecule of EGFR, predict clinical response to EGFR inhibitors, but this has yet to be validated in a larger number of cases.[157] Notably, a single substitution of glutamic acid for valine at codon 600 (V600E) accounts for approximately 90% of the BRAF missense mutations found in human tumors.[159] Considering BRAF is a serine/threonine kinase that is commonly activated by a somatic point mutation in human cancer, it may provide new therapeutic opportunities in a subset of NSCLC.
ALK Rearrangement ALK encodes a TK receptor found in a number of fusion proteins consisting of the intracellular kinase domain of ALK and the amino terminal portions of different genes.[166,167] A subset of NSCLC cases harbor a transforming fusion gene, EML4–ALK, within the genome. To date, seven gene fusion variants have been reported in NSCLCs. All involve the intracellular TK domain of ALK starting at a portion encoded by exon 20.[168] This fusion is formed as the result of a small inversion within the short arm of chromosome 2 that joins EML4 to ALK, inv(2)(p21;p23), which encodes an activated TK protein.[169,170] Several other transforming EML4–ALK fusion gene variants have also been identified, involving various EML4 exons and ALK.[171,172] ALK can also fuse with some other rare fusion partners, such as KIF5B and TFG.[173]
Activated ALK is involved in the inhibition of apoptosis and the promotion of cellular proliferation through activation of downstream PI3K–AKT and MAPK signaling pathways.[174] The EML4–ALK fusion is a rare abnormality detected in approximately 6% of patients with NSCLC.[144,169,175] Fusion of the EML4–ALK gene and its associated EML4–ALK product may lead to constitutive activation of the RAS–RAF–MEK–MAPK pathway.[47] In addition, two other less frequent ALK fusions in lung cancer have been reported.[176]
Non-small-cell lung cancer cases harboring EML4–ALK are characteristically found to have wild-type EGFR, as well as wild-type KRAS.[177,178] In addition, patients with these tumors tend to be younger, have an advanced clinical stage at presentation, have never smoked and their tumors exhibit solid histology, often with a component of signet ring cells.[178,179]
Patients with this alteration demonstrated an extraordinary response to the MET–ALK inhibitor, PF-02341066, in a Phase I/II trial.[144] Ten out of 19 patients (53%) experienced objective responses. A total of 15 out of 19 patients (79%) demonstrated disease control at 8 weeks, lasting as long as 40 weeks in some. Only four patients demonstrated disease progression. Kwak et al. screened 1500 NSCLC patients and identified 82 patients with advanced ALK-positive disease.[180] The vast majority (96%) of these cancers were adenocarcinomas. A total of 94% of these patients had received at least one prior therapy. All of these patients were treated with crizotinib, an oral small-molecule ALK TKI. The disease control rate was 90%, which included a 57% response rate plus 33% with stable disease. Furthermore, Rodig et al. evaluated the incidence and the characteristics of ALK-rearranged lung adenocarcinomas within the western population to elucidate the optimal diagnostic modality to detect ALK rearrangements in routine clinical practice.[179] In their study, 358 lung adenocarcinomas were tested for ALK rearrangement by FISH and immunohistochemistry. ALK rearrangement again demonstrated an association with younger age, never having smoked, advanced clinical stage, and a solid histology with signet ring cells in some cases. Of note, none of the ALK-rearranged tumors harbored coexisting EGFR mutations. The results of this study demonstrate that ALK rearrangements are uncommon in the western population, but represent a distinct clinical entity with unique attributes and the possibility of distinct treatment options. For suspected cases, dual diagnostic testing with FISH and immunohistochemistry should be considered in order to accurately identify lung adenocarcinomas with ALK rearrangement.[179] A study by Zhang et al. involving 266 Chinese patients with NSCLC revealed approximately similar results.[178]
Shaw and colleagues investigated 141 NSCLC cases and found that 19 (13%) contained EML4–ALK fusion, 31 (22%) demonstrated EGFR mutantations and 91 (65%) were wild-type for both.[144] Several studies provide consistent evidence that EML4–ALK and EGFR mutations are mutually exclusive.[144,169,177,181] However, Tiseo et al. reported a 48-year-old Caucasian man who had never smoked and who was diagnosed with NSCLC with a concomitant EGFR mutation and ALK translocation that was resistant to erlotinib therapy.[182] This may be representative of the nature of this subgroup of NSCLC.[154]
No EGFR mutations in the EML4–ALK cohort and no instances of ALK rearrangement in the EGFR cohort have been found.[144] Among patients with metastatic disease, EML4–ALK rearrangements were associated with resistance to EGFR TKI treatment.
Compared with the EGFR-mutated and wild-type EGFR/ALK cohorts, patients with EML4–ALK fusion gene-positive tumors were significantly younger and more likely to be male.[144] However, another study demonstrated that males and females are equally affected.[139] Patients with EML4–ALK-positive tumors are more likely to be never/light smokers. A total of 18 of the 19 EML4–ALK-positive tumors were adenocarcinomas, predominantly of the signet ring cell subtype.[144] EML4–ALK defines a molecular subset of NSCLC with distinct clinical characteristics. Specifically, patients who harbor this mutation do not benefit from EGFR TKIs and should be directed to trials investigating ALK-targeted agents.
Summary
Two classes of anti-EGFR agents – monoclonal anti-EGFR antibodies and small-molecule EGFR TKIs – are currently used in EGFR-targeted therapy.
Additional mutations in EGFR are an important mechanism of acquired resistance to EGFR-targeted therapy.
Some mutations, such as exon 20 D770–N771 and T790M, are associated with EGFR TKI resistance.
Oncogenic activation of EGFR downstream effectors, such as KRAS, BRAF, PIK3CA and deletion of PTEN, is associated with resistance to TKI therapy.
KRAS mutation accounts for approximately 35–45% of TKI-nonresponsive NSCLC patients.
KRAS and BRAF mutations, as well as ALK rearrangements, are mutually exclusive with EGFR mutations.
ALK rearrangements are more common in younger patients who have never smoked, and their tumors often exhibit solid architecture, signet ring cell histology and wild-type EGFR and KRAS.
Conclusion
The EGFR is an effective therapeutic target in treating NSCLC. EGFR mutations have been used as a selection criterion for EGFR TKIs and are also used as a predictive marker for responsiveness to EGFR-targeted therapy. However, evidence is beginning to demonstrate that NSCLC may be composed of multiple subsets of tumors, each with its own molecular abnormalities.
Identification of the relevant molecular subtypes of this heterogeneous disease and selecting patients for the appropriate targeting agents is critical in the personalized therapy of NSCLC. KRAS/BRAF mutations, which are mutually exclusive with EGFR mutations, are rare in NSCLC but may be important mechanisms in the etiology and prediction of resistance to EGFR TKI therapy.
In conclusion, one unifying predictive model does not apply to all tumor types, and the larger goal of discovering a predictive marker to guide patient selection for EGFR-targeted therapy remains elusive. EGFR alteration markers need to be further evaluated in combination with clinical data to provide clear rationales for future therapeutic strategies in the treatment of NSCLC.
|
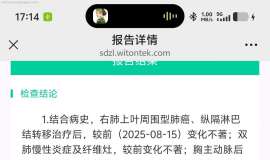
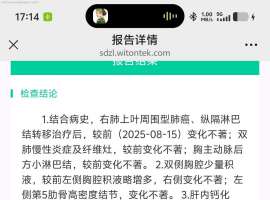 不知不觉进入第七年。。胸腔积液怎么
中间这几年很顺利也很稳定 这几年都没再关注这个平台。空窗一年半,最近一次复查胸腔
不知不觉进入第七年。。胸腔积液怎么
中间这几年很顺利也很稳定 这几年都没再关注这个平台。空窗一年半,最近一次复查胸腔
 扫清治疗迷雾!赵军教授、刘晓梅教授
作者:雨过天晴“一代药物耐药后,能直接换成新一代药物吗?”“复查发现脑部有几个很
扫清治疗迷雾!赵军教授、刘晓梅教授
作者:雨过天晴“一代药物耐药后,能直接换成新一代药物吗?”“复查发现脑部有几个很
 发生率高达97%!抗癌路上,别让口腔
作者:雨过天晴
口腔黏膜炎作为癌症患者在抗肿瘤治疗过程中最常见的并发症之一,其高
发生率高达97%!抗癌路上,别让口腔
作者:雨过天晴
口腔黏膜炎作为癌症患者在抗肿瘤治疗过程中最常见的并发症之一,其高
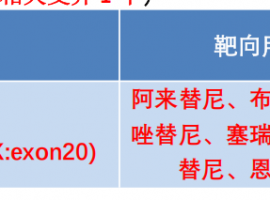 爸爸晚期肺癌肿瘤已经看不到了!我确
讲述者:陈先生整理者:pear
适逢暑假得闲,我带着全家自驾出游,正将最后一件行李塞
爸爸晚期肺癌肿瘤已经看不到了!我确
讲述者:陈先生整理者:pear
适逢暑假得闲,我带着全家自驾出游,正将最后一件行李塞
 肺腺癌10年,伏美替尼 心血管副作用
奶奶78岁,肺腺癌10年,2017.7-2023.7第一代靶向药凯美纳6年,2023.7至今伏美替尼1年
肺腺癌10年,伏美替尼 心血管副作用
奶奶78岁,肺腺癌10年,2017.7-2023.7第一代靶向药凯美纳6年,2023.7至今伏美替尼1年





 显身卡
显身卡