"Pan inhibitors permanently and irreversibly stop certain functioning of EGFR. Initial cell studies have indicated these stronger inhibitors can work against the resistant cells with the mutation. To determine whether the T790M mutation leads to resistance to EGFR inhibitors that have different molecular structures and mechanisms, we screened four commercially available EGFR inhibitors (AG1478, cetuximab, erlotinib, and CL-387,785) using cells that were transiently transfected with the delL747–S752 construct and the delL747–S752+ T790M construct. We consistently found that CL-387,785, a specific and irreversible anilinoquinazoline EGFR inhibitor, strongly inhibited EGF-induced phosphorylation While this may not be a valid alternative for many patients, it may make sense for patients whose cancers have been shown to be associated with EGFR."( S" N& f" b* H- V, x+ m& f/ x* K
( k+ M/ s1 I( G+ X"Some recurrent tumors have a common secondary mutation in the EGFR kinase domain, T790M, conferring drug resistance, but in other cases the mechanism underlying acquired resistance is unknown. In studying multiple sites of recurrent NSCLCs, we detected T790M in only a small percentage of tumor cells.... Although gefitinib-resistant clones are cross-resistant to related anilinoquinazolines, they demonstrate sensitivity to a class of irreversible inhibitors of EGFR. These inhibitors also show effective inhibition of signaling by T790M-mutant EGFR and killing of NSCLC cells with the T790M mutation." Kubayashi9 e0 h' B, j7 ^. ~8 @4 [- L
! J$ \+ E& A+ V8 M4 [
Its success in human studies has been debatable. There have been several trials but none sufficient impressive to move towards FDA approval. Many of these studies did not deal solely with T790M but a variety of patients. Part of the problem may be the company's desire to secure aa drug that is effective overall, and reports testing for the T790M mutation and detailing the drug's impact are strangely difficult to find.
8 a4 Q, A6 Z1 ^# ]2 t6 A( O+ G# m; a) M( v- Z0 T8 n
) b) w5 m# |* l( TB. HKI 272( }$ v% a! x( ^( o
! O% z9 R+ R: H, D
8 P) r6 B; T& J2 x: U
One cell study found pan-inhibitor HKI 272 effective with tumor cells in a laboratory setting. "HKI 272 is effective in inhibiting various lung cancer mutations- "HKI-272 is effective in growth inhibition of Ba/F3 cells transformed with EGFRvIII, EGFR-L858R, and EGFR-L858R-T790M." Ji (1)* H$ E2 q6 e p* P
9 {6 g) R; h$ z( w9 J+ N1 O5 s/ j/ b% N) r# w9 [: p. J
C. Lapanatib
: s; D6 V# W' y2 ^0 G4 f
; _3 X' j; `/ n/ G4 F; ZA recent study showed promise for a combination of Cetuximab (Erbitux) and Lapatanib (Tykerp),
L6 K4 k$ q/ e
8 s: B6 L& K3 I$ C }' E! Q7 v
8 |5 Q1 I2 Y. k# j' O5 _: Q& P# k' O2 D$ _! t' \# t8 U
"In this study, we show that a combination of lapatinib and cetuximab overcomes gefitinib resistance in NSCLC with the T790M mutation. We observed that T790M lung cancer cells were resistant to gefitinib, and Stat3 was persistently activated in the resistant cells. A reversible EGFR and HER2 TKI, lapatinib, decreased Stat3 activation by blocking heterodimerization of EGFR and HER2, which led to a modest increase in the inhibitory effect on gefitinib-resistant T790M cells. In addition to lapatinib, the anti-EGFR antibody, cetuximab, induced down-regulation of EGFR and apoptotic cell death in T790M cells. Finally, combined lapatinib and cetuximab treatment resulted in significantly enhanced cytotoxicity against gefitinib-resistant T790M cells in vitro and in vivo. Taken together, these data suggest that treatment with a combination of lapatinib and cetuximab, which induces dimeric dissociation and EGFR down-regulation, appears to be an effective strategy for treatment of patients with EGFR TKI-resistant NSCLC."
9 t& V) \2 x: S8 |
3 Q# f3 v+ ^$ n6 t/ o
& i5 I! ^; J ?. W9 ZBoth Erbitux and Lapatanib are FDA approved drugs, though not specifically approved for this purpose. They can be prescribed off label if a physician chooses. ?$ R5 \+ y3 l S* X2 ?% v
! x- \1 P$ p( q& k0 r, Z* s
D. Zactima (ZD-6474)3 P$ d+ T% g- R, r$ m9 U, ?
! b0 ~8 a9 X- M4 e' m8 W
+ H* ?8 }! k X {4 O
E. BMS 690514
* @4 W3 R9 Y- J2 s% w% x6 o$ q3 I6 d! P) M; ?0 ?
Only cell studies are available. "BMS-690514, a novel panHER/vascular endothelial growth factor receptor (VEGFR) inhibitor described here, exerted antiproliferative and proapoptotic effects on NSCLC cell lines, with prominent efficacy on H1975 cells expressing the T790M mutation.", r( v7 L/ y" P% B! q# O0 B
) ?6 q4 [$ j& V8 L. m' w0 r7 z4 |
|
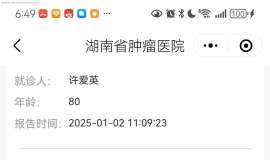
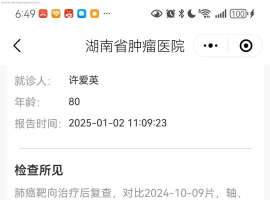 求助,请大家帮忙出出主意,奥西+280
病人是我妈妈,43年8月的,81周岁。
19年10月28日确诊,肺腺癌晚期,右上肺肺癌并双
求助,请大家帮忙出出主意,奥西+280
病人是我妈妈,43年8月的,81周岁。
19年10月28日确诊,肺腺癌晚期,右上肺肺癌并双
 求助:肺腺癌3年,脑转脑膜转骨转,E
肺腺癌3年,脑转脑膜转、骨转,E19+797S突变,免疫阴性,阿美29个月,期间放疗2次,后
求助:肺腺癌3年,脑转脑膜转骨转,E
肺腺癌3年,脑转脑膜转、骨转,E19+797S突变,免疫阴性,阿美29个月,期间放疗2次,后
 父亲晚期肺癌即将迈入第7年,这是我
讲述者:郭先生整理者:pear
2018年4月底,父亲因长期右肩胛骨疼痛、脸部及颈部肿大而
父亲晚期肺癌即将迈入第7年,这是我
讲述者:郭先生整理者:pear
2018年4月底,父亲因长期右肩胛骨疼痛、脸部及颈部肿大而
 妈妈与脑膜转奋战4年多,如今也成功
作者:李妮妮
2020年12月1号,妈妈在确诊晚期肺癌4年零10月,经历了化疗、一代靶向易
妈妈与脑膜转奋战4年多,如今也成功
作者:李妮妮
2020年12月1号,妈妈在确诊晚期肺癌4年零10月,经历了化疗、一代靶向易
 ret错义突变可以用普拉替尼吗?
父亲肺鳞癌,无法手术。医院的基因检测报告上提示:肺鳞癌RET基因(NM_020975.4) 3号外
ret错义突变可以用普拉替尼吗?
父亲肺鳞癌,无法手术。医院的基因检测报告上提示:肺鳞癌RET基因(NM_020975.4) 3号外






 显身卡
显身卡

 高高兴兴是一天,愁眉苦脸还是一天,就看如何选择。生命会随着时间的流失而慢慢远去的,珍惜现在的每一天,过好每一天是最重要的,别给自己吃后悔药。
高高兴兴是一天,愁眉苦脸还是一天,就看如何选择。生命会随着时间的流失而慢慢远去的,珍惜现在的每一天,过好每一天是最重要的,别给自己吃后悔药。